Claudio Della Volpe
L’episodio di Mestre ha messo sotto i riflettori la possibilità di incendio dei mezzi alimentati elettricamente; e dunque ha senso discutere un po’ questo problema basandoci sui dati e sulla chimica che conosciamo.
Partiamo dalle probabilità reali di incendio dei mezzi elettrici. Dato che i mezzi elettrici sono poco diffusi non ha senso paragonare il numero assoluto di incendi su mezzi elettrici o fossili ma occorre paragonare le percentuali di incendio rapportandole al numero di mezzi in circolazione; è pur vero che certe tipologie di incidente saranno poco probabili, per esempio lo scontro diretto fra due mezzi elettrici è altamente improbabile al momento.
Se si usano i dati dei paesi con statistiche affidabili e con il maggior numero di mezzi elettrici: USA, Svezia, Norvegia, Australia si trova che le probabilità di incendio sono molto più basse rispetto alla probabilità di incendio per i mezzi fossili di qualunque tipo. Purtroppo dati analoghi in Cina che è il paese col maggior numero di EV non ce ne sono[1].
Parecchi dati vengono dalle assicurazioni che hanno ovviamente interesse a valori credibili; e proprio da questi viene fuori che c’è uno o due ordini di grandezza di differenza o più nella percentuale di incendi fra EV e fossili di qualunque tipo.

Dati validi per gli USA nell’anno giugno 22-giugno 23 da documenti citati in nota 1.
Pare inoltre da questi dati che i veicoli peggiori siano gli ibridi mentre i più sicuri sarebbero proprio gli elettrici.
Risultati analoghi o perfino più favorevoli agli EV sono riportati per gli altri paesi occidentali.
Ma al di là dei numeri, che probabilmente diventeranno più precisi in futuro col crescere del ruolo della mobilità elettrica, quali sono i meccanismi degli incendi e quali sono i rischi connessi e i metodi antincendio migliori?
La cosa basilare da considerare è che una batteria è un ambiente in cui si sviluppa in forma elettrochimica una normale reazione chimica che mette in gioco flussi significativi di energia e cambia i numeri di ossidazione dei reagenti partecipanti; tale energia viene sfruttata in forma elettrica ma è anche possibile convertirla direttamente in calore.
A differenza di una reazione normale in cui gli elettroni vengono scambiati direttamente fra i reagenti, in elettrochimica gli elettroni vengono scambiati SOLO agli elettrodi, una sorta di mercato controllato non libero, per fare una analogia economica, quasi un socialismo elettronico.
Ma nonostante tale forma ordinata di processo non tutta l’energia libera può trasformarsi in elettricità: è il famoso secondo principio della termodinamica; una parte diventa comunque calore, scambio incontrollato di energia.
L’elemento critico per regolare questo parametro è la velocità di reazione.
Non è possibile trasformare direttamente in energia elettrica tutta la variazione di energia libera della reazione, una quota di tale trasformazione anche nel caso migliore diventa calore; si può porre uguale al calore la quota di energia persa per effetto Joule e legata alla corrente: più alta è la corrente, che misura direttamente la velocità di reazione, maggiore è la trasformazione in calore; teoricamente si può avere la massima trasformazione in energia elettrica operando a velocità molto bassa ossia con correnti molto basse al limite nulle; più cresce la velocità ossia la corrente più cresce il calore e diminuisce la quota di elettricità.
Ovviamente nelle batterie reali occorre accettare questo criterio e scegliere una situazione di compromesso, la velocità di reazione che ci serve, ossia la corrente e dunque la potenza che vogliamo e la corrispondente dispersione di calore.
Si capisce dunque che un po’ di calore deve essere sempre dissipato e a volte questo è perfino positivo per il processo elettrochimico contribuendo al mantenimento della temperatura ottimale.
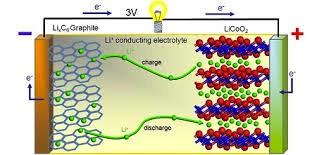
Nel caso delle batterie al litio ricaricabili uno ione litio può occupare due stati energetici diversi, uno nel materiale dell’anodo e uno nel materiale del catodo.
Quando si carica la batteria si sposta lo ione litio e il corrispondente elettrone in un contenitore prevalentemente di carbonio grafitico, fra due strati di grafene diciamo, un fenomeno che prende il nome di intercalazione, lo ione litio viaggia in soluzione di opportuno solvente, spesso polimerico ma non sempre e l’elettrone nel filo di rame.
Nell’anodo lo ione litio ha potenziale energetico più alto incapsulato fra due esagoni di atomi di grafene interagendo con i suoi elettroni pi-greco e il suo elettrone; quando si scarica la batteria il litio ione diffonde nell’elettrolita tornando nel suo contenitore a più bassa energia, comunemente un ossido misto di litio e di altri elementi: cobalto, manganese o altri composti come il fosfato di ferro, mentre l’elettrone fluisce nel cavo facendo lavoro elettrico e calore .
In nessun caso abbiamo a che fare con litio metallico, ma sono comunque due stati “di ossidazione” diversi del sistema Li-grafite o Li in ossido misto o Li-ferro fosfato, come se cambiassero numero di ossidazione il cobalto e il carbonio o il ferro oltre che il litio medesimo.
Il salto energetico ottenuto in questo modo è fra i più alti ottenibili, differenze di potenziale di oltre 4V possono essere gestite con successo. 4V per una mole di elettroni (quasi 100mila coulomb) corrispondono a qualcosa come (ricordiamo che J=VxC) 400 kilojoule/mole!
Ovviamente non possiamo usare un solvente come l’acqua perché a poco meno di 2 volt l’acqua si distrugge trasformandosi nei suoi componenti; dobbiamo usare altri solventi sempre in grado di sciogliere ioni ma non così instabili; i migliori sono polimeri come il PEO; e ovviamente abbiamo bisogno di controioni negativi per bilanciare le cariche del litio (la elettroneutralità è una condizione di esistenza dei sistemi elettrochimici!); anche questi ioni devono essere stabili ma assicurare grande conducibilità elettrica; le specie più adatte spesso contengono fluoro, per esempio lo ione PF6–, esafluorofosfato.
Tutte queste sostanze sono stabili nelle condizioni di lavoro comuni, ma possono perdere di stabilità come farebbe l’acqua se tali condizioni cambiano.
Data la grande quantità di energia in gioco questo può succedere; per esempio se la quota di dissipazione aumenta fornendo più calore e dunque aumentando la temperatura del sistema i vari componenti diventano instabili.

Il componente grafitico può bruciare se riscaldato in presenza di ossigeno producendo uno degli ossidi di carbonio; ma attenzione lo stesso possono fare quei leganti polimerici che vengono usati per tenere insieme una struttura micro o nanometrica delle particelle attive degli elettrodi (si tratta di numerosi polimeri analizzati in letteratura[2] i polimeri sono sparsi a piene mani nelle strutture della batteria al litio (LIB)); ma da dove viene l’ossigeno? Può venire da due fonti una esterna, se la batteria, comunemente isolata dall’ambiente esterno, viene forata oppure se un aumento di temperatura esterno è in grado di destabilizzare il catodo; gli ossidi misti in queste condizioni possono liberare ossigeno gassoso; anche il polimero che funge da elettrolita può bruciare, reagire con ossigeno ed infine l’esafluorofosfato può liberare acido fluoridrico che è un gas molto aggressivo (corrode anche i vetri).
La sorgente più comune di calore è un corto circuito fra due elementi della batteria, l’anodo e il catodo, che può dipendere da una deformazione della batteria, dovuta per esempio ad un urto violento.
Un corto circuito è una condizione in cui TUTTA l’energia libera disponibile diventa calore e la quota di lavoro elettrico si azzera.
Le batterie al litio hanno una tipica struttura a spirale o comunque multistrato per accrescere la superficie di reazione; dunque una sua deformazione anche di piccola entità può produrre questo effetto.
Data questa situazione ogni occasione di surriscaldamento della batteria è rischiosa; per esempio la carica della batteria, specie se veloce può compromettere una batteria mal costruita o che abbia subito un precedente danno meccanico; stiamo parlando di spessori degli strati di decine di micron e dunque piccole deformazioni sono critiche.
Aggiungiamo un’ultima considerazione: i processi di cui abbiamo parlato diventano di fatto autocatalitici: il calore li stimola ed essi producono calore, una retroazione positiva; questo ha portato alla definizone comune di “thermal runaway”.
Ovviamente queste cose sono risapute e le batterie sono costruite tenendone conto (ciononostante ricorderete sicuramente il caso delle batterie del Samsung Note7 che bloccò la commercializzazione del dispositivo mettendo in crisi Samsung).
Anche se le batterie al litio sono piano-parallele ossia gli elettrodi piani sono separati a loro volta da uno strato conduttore di spessore costante, il tutto è avvolto in forma di cilindro, di esagono, di parallelepipedo inserito in un contenitore plastico o metallico; l’uso di centinaia o migliaia di batterie sui dispositivi maggiori come i motori d’auto comporta che la dissipazione di calore può avere effetti diversi e può essere dissipato in modo più o meno facile; per esempio la produzione di gas in un contenitore rigido può causare una esplosione oppure l’espansione del contenitore plastico con ulteriori deformazioni. E il calore prodotto viene dissipato con opportuni apparati o usato perfino per scopi vari.
Un Battery Management System (BMS), controlla il funzionamento del tutto che è costruito collegando le batterie in modo furbo che consente all’elettronica del BMS di evidenziare anomalie in singole batterie o gruppi di batterie, per poterle escludere o sostituire e di regolare la velocità di ricarica.
Consigli banali possono essere di usare sempre batterie di qualità (le batterie sono prodotte in gran numero e in una gaussiana di qualità ci saranno sempre batterie più o meno “buone”; caricarle con dispositivi anch’essi progettati dal costruttore, originali, e non lasciarle mai senza un controllo sia pur qualitativo che ne evidenzi le deformazioni (una batteria che si rigonfia casomai sposta la meccanica del vostro dispositivo: cellulare, computer, il touchpad o la tastiera ne risentono, la forma del cellulare si altera, etc) o la temperatura eccessiva.
Su miliardi di batterie qualcuna potrà sempre surriscaldarsi o perfino esplodere; il rischio si può ridurre ma non azzerare; d’altronde l’incendio di fossili è talmente diffuso nel nostro mondo che abbiamo istituito da secoli un corpo di professionisti i Vigili del Fuoco per gestire questo rischio; e i Vigili del Fuoco si aggiornano in continuazione.
A questo riguardo occorre ricordare che l’incendio delle batteria NON è un incendio di tipo D, da combustione di metalli (polveri di litio, magnesio, alluminio) ma è classificato come incendio nelle classi A e principalmente B, combustione di solidi e liquidi e come tale deve essere trattato; l’acqua (in grandi quantità, circa 10 volte maggiori che nell’incendio tradizionale) o la polvere di sostanze fluorurate o la CO2 possono essere usati; quest’ultima tende a raffreddare molto essendo fornita in stato condensato e dunque assorbendo energia per evaporare oltre che soffocando l’incendio.

Ancora da notare che il comportamento dell’incendio di LIB è più insidioso perché rispetto ad uno tradizionale l’inizio della fiamma libera può coincidere con l’esplosione se i contenitori sono rigidi e dunque con una fase di incontrollabilità, mentre questa fase è più lenta da raggiungere negli incendi tradizionali Comunque data la complessità dei sistemi usati un incendio di batteria al litio dura più di un incendio comune ed esige dispositivi e spazi adeguati, come depositi dove trasportare il mezzo che brucia anche se l’incendio è terminato in apparenza; potrebbe riprendere; un maggior numero di estintori deputati ad incendi di questo genere, le polveri classiche possono non bastare, quelle alogenate sono meglio; insomma occorre imparare ad affrontarli. Al momento alcuni grandi carrier marittimi evitano di trasportare le batterie; mentre il settore critico che ha usato sempre più LIB è paradossalmente l’aeronautica a causa del più favorevole rapporto peso-potenza, ma dove un incendio od una anomalia può essere fatale.
Tocca comunque ribadire quel che abbiamo detto all’inizio; le statistiche smentiscono il maggior rischio di incendio delle batterie rispetto alle sorgenti fossili; se si fa riferimento al numero di impianti o di auto circolanti gli incendi sono si possibili ma in quantità enormemente inferiore; i Vigili del fuoco sono consci delle differenze sulla tipologia di fuoco; casomai sono da aggiornare le tipologie di estintori da usare e i corsi di formazione dei volontari dovranno contenere anche questo tema, con crescente importanza.
[2] https://www.sciencedirect.com/science/article/abs/pii/S0360128519302138
Documenti consultati.
1-A review of lithium-ion battery safety concerns: The issues, strategies, and testing standards
Journal of Energy Chemistry 59 (2021) 83–99
2 – Procedia Engineering 211 (2018) 629–634 Procedia Engineering 00 (2017) 000–000
Experimental Study on fire and explosion suppression of self ignition of Lithium Ion Battery
3 – Thermal Stability Studies of Li-Ion Cells and Components
Journal of The Electrochemical Society, 146 (9) 3224-3229 (1999) S0013-4651(99)02-009-1
Thermal Stability Studies of Li-Ion Cells and Components Hossein Maleki, Guoping Deng,* Anaba Anani, and Jason Howard